News & events:
-
Felix Concordia API for NET 9
C# Fc-API provides FAST performances on Linux and Windows
Originally developed for the .NET Framework, our Fc-API Advanced Programming Interface is now available for cross-platform .NET 9 (formerly .NET Core). This free and open-source software framework, compatible with Windows, Linux, and macOS, allows you to integrate our Felix Concordia cheminformatics and bioinformatics classes, methods, and functions into any new custom-developed application.
For a Felix Concordia client, we implemented our Fast Substructure Search (FcFastSubstructureSearchFingerPrintEngine class) in their software application on Linux and Windows servers, achieving a throughput of nearly one million 2D molecular substructure comparisons per second per CPU core (E5-2600 Xeon processor). A 40-core motherboard with hyperthreading offers 80 million comparisons per second.
-
Web interfaces
Accessing FcLigand features through a web browser:
The performance of calculations and graphical rendering was evaluated in a client-server web architecture using the Blazor framework (open-source framework from Microsoft to create web user interfaces based on components using C# and HTML). Blazor leverages WebAssembly technology developed by the World Wide Web Consortium since 2017. It delivers top-tier performance with a near-native instruction set for both x64 and ARM processors.
Based on this successful validation using unified C# code, we are now working on delivering full web-based graphical interfaces for FcBioisostere, FcLigand, and FcRebuilder software.
-
Our commitment to Corporate Social Responsibility in 2023
Promoting women’s rights with Cité des Droits des Femmes association:
In 2023 we were proud to initiate activities at Felix Concordia to promote human rights, which is one of the seven core subjects of the ISO 26000 international standard for social responsibility, a CSR guideline.
We became the first corporate sponsor of the Cité des Droits des Femmes project (https://CiteDesDroitsDesFemmes.fr), which aims to open the first museum in the Paris region dedicated to the history of women’s rights. Social rights in the workplace is one of their key themes.
A recent initiative was an interactive photo exhibition featuring slogans from International Women’s Rights Day, covering topics ranging from violence against women to gender equality in the workplace.

-
Enhancements on Bioisosteric replacement
FcBioisostere new graphical features to better explore bioisosteric PDB based pairs:
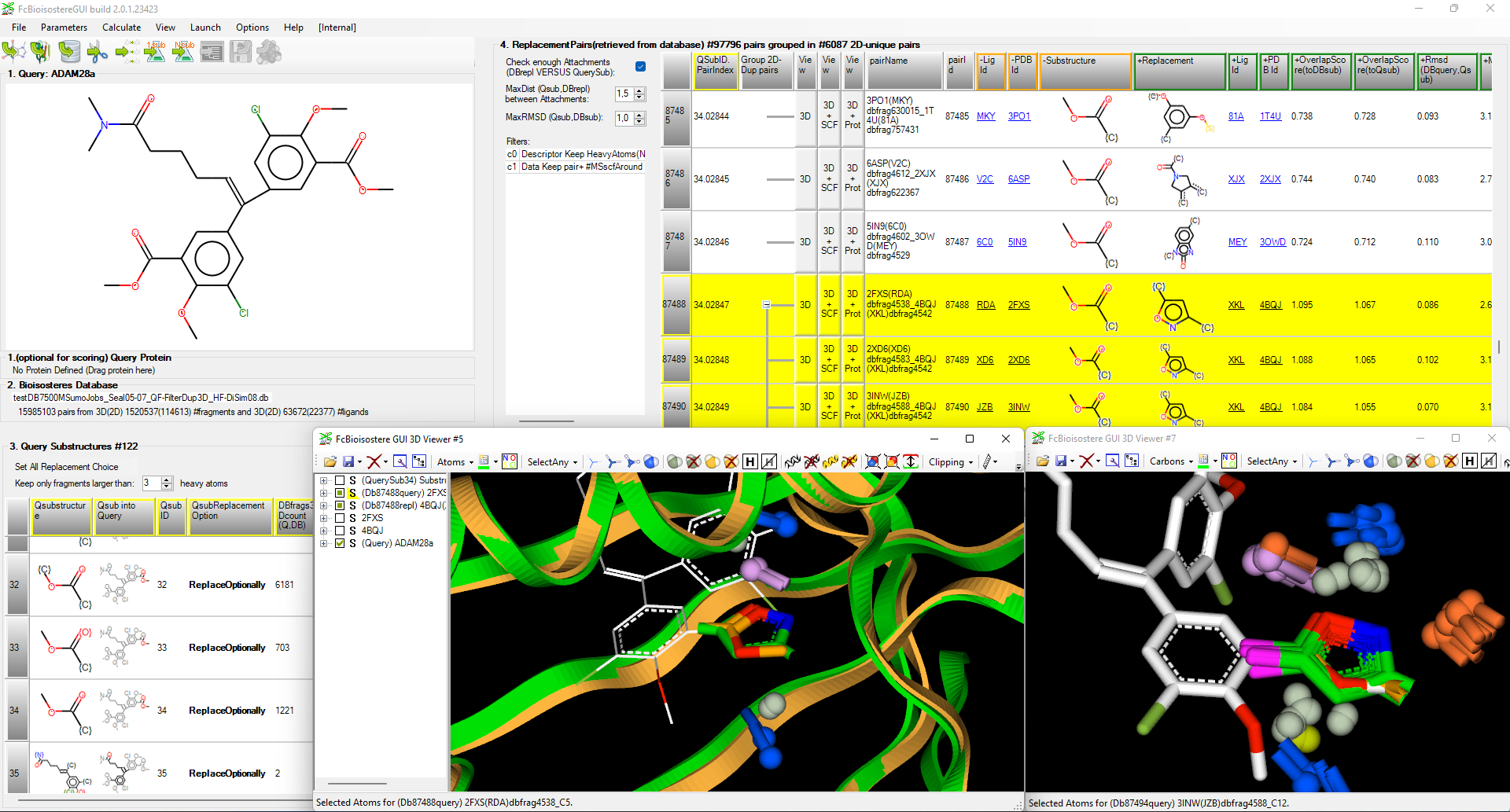
This year we extanded the FcBioisostere software to better sort the precalculated 3D bioisosteric pairs extracted by our application from the PDB Protein Data Bank. Same topological pairs are now grouped together to help users assess a statistical suitability of the proposed fragment replacement onto their ligand of interest. Furthermore, Structural Chemical Features (Hbond donor/acceptor, charge, hydrophobicity, …) and protein of proposed bioisosteric pair can be displayed alongside the input ligand.
These and other user enhancements are now available in the latest FcBioisostere release.
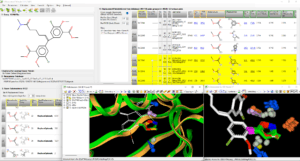
Fig.: Replacement of hydrolysable methyl ester chemical moiety on ADAMs ligand query (pdb code 3SI9) with FcBioisostere ; on the left, upper view shows the input 2D or 3D query, lower table shows all query substructures to select the area to replace ; on the right, upper table shows all found replacement pairs, the selected pair is yellow was detected in 121 PDB pairs, lower view shows first the query ligand in white with the protein-fragment pairs in green and orange, then on the right, some of the 121 pairs of methyl-ester by 1,2-oxazole replacement.
-
Introduction to FcPipeline
Designing protocols with a workflow user interface:
In order to faster design new protocols for users in our C2P Chemo-Proteomic Platform, we investigated technical solutions to provide a sketcher to users to design predictive protocols similar to a workflow.
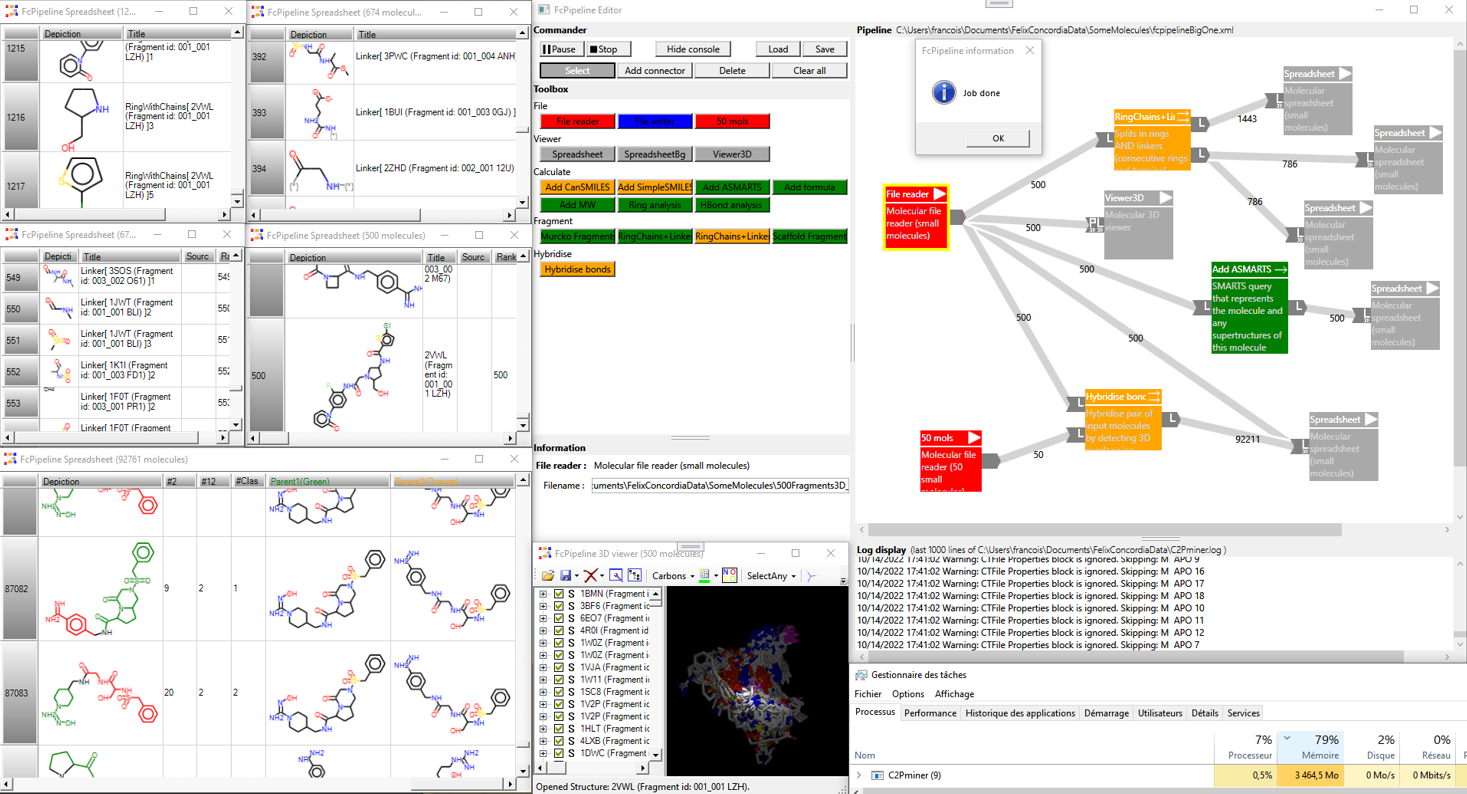
FcPipeline our new module under development provides a full set of input/calculation/output nodes that can be chained together to define any chemoproteomic protocols. The user interface dynamically displays the number of processed molecules at each stage of the workflow. The processing of ligand and protein flows is managed using iterator technology, which allows for immediate results for a given protocol, enabling users to verify the protocol’s validity without having to wait for the entire calculation to finish.
Any of our C2P API methods can be integrated as a separate node, with parallel processing enabled for that node if needed to improve performance. We plan to include all our chemo-proteomics C# methods in a future release of FcPipeline.
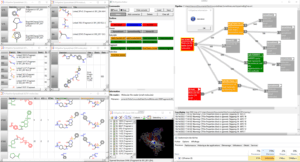 Fig.: FcPipeline protocol for fragmenting and 3D-hybridizing two sets of pre-superimposed ligands; red nodes represent input readers; orange and green nodes represent cheminformatics methods; gray nodes are viewers (all displayed on the left side of the view, with the 3D hybrid structures shown below, using red or green substructures to indicate the source ligand)
Fig.: FcPipeline protocol for fragmenting and 3D-hybridizing two sets of pre-superimposed ligands; red nodes represent input readers; orange and green nodes represent cheminformatics methods; gray nodes are viewers (all displayed on the left side of the view, with the 3D hybrid structures shown below, using red or green substructures to indicate the source ligand)
-
Working on COVID-19
Proof Of Concepts to annotate protein surfaces and generate drug candidates:
Since the beginning of the COVID-19 pandemic crisis, the scientific community has generated an amazing amount of data especially in structural biology with more proteins together with cocrystallised ligands of fragment probes. We ran our 3D intermolecular surface miner protocol in to first superpose the PDB materials having similar molecular surface, then combined in FcLigand the superpose ligand/fragment materials into protein pockets, and finally annotated the generated molecules by matching to PDB/Pubchem/ChEMBL repositories.
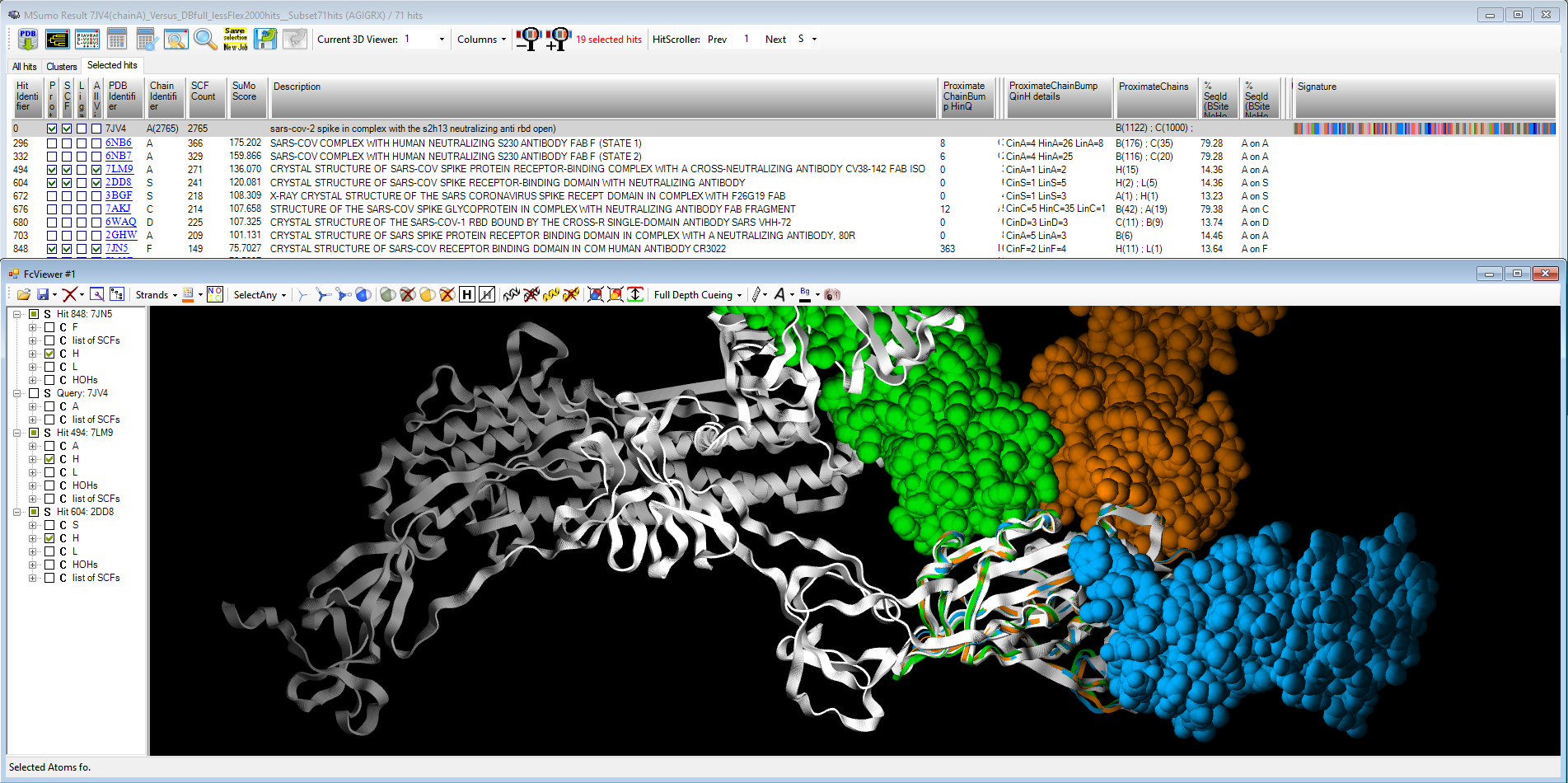
On Spike protein that project from SARS-CoV-2 surface (pdb code 7JV4), we compared the full 3D surface to retrieve PDB proteins showing similar local network of 3D interactions. The resulting superposition highlights epitopes positions on the RBD Rigid Body Domains targeted by actual vaccines.
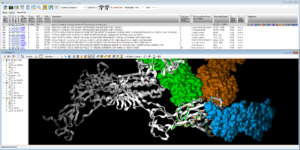 Fig. Epitope detection on SARS-CoV-2 Spike protein: spreadsheet on top summarizes 19 non-SARS-CoV-2 proteins that interact with Spike protein ; 3D viewer displays in white the 7JV4 query backbone and 3 hits in ball rendering, n°494 (7LM9 in blue), n°604 (2DD8 in orange), and n°848 (7JN5 in green vert) ; only antibody part of hits are displayed, network of 3D shared structural chemical features (Hbond donor/acceptor, charge, hydrophobicity, aromatic, …) for the 3 hits highlights putative epitope surfaces.
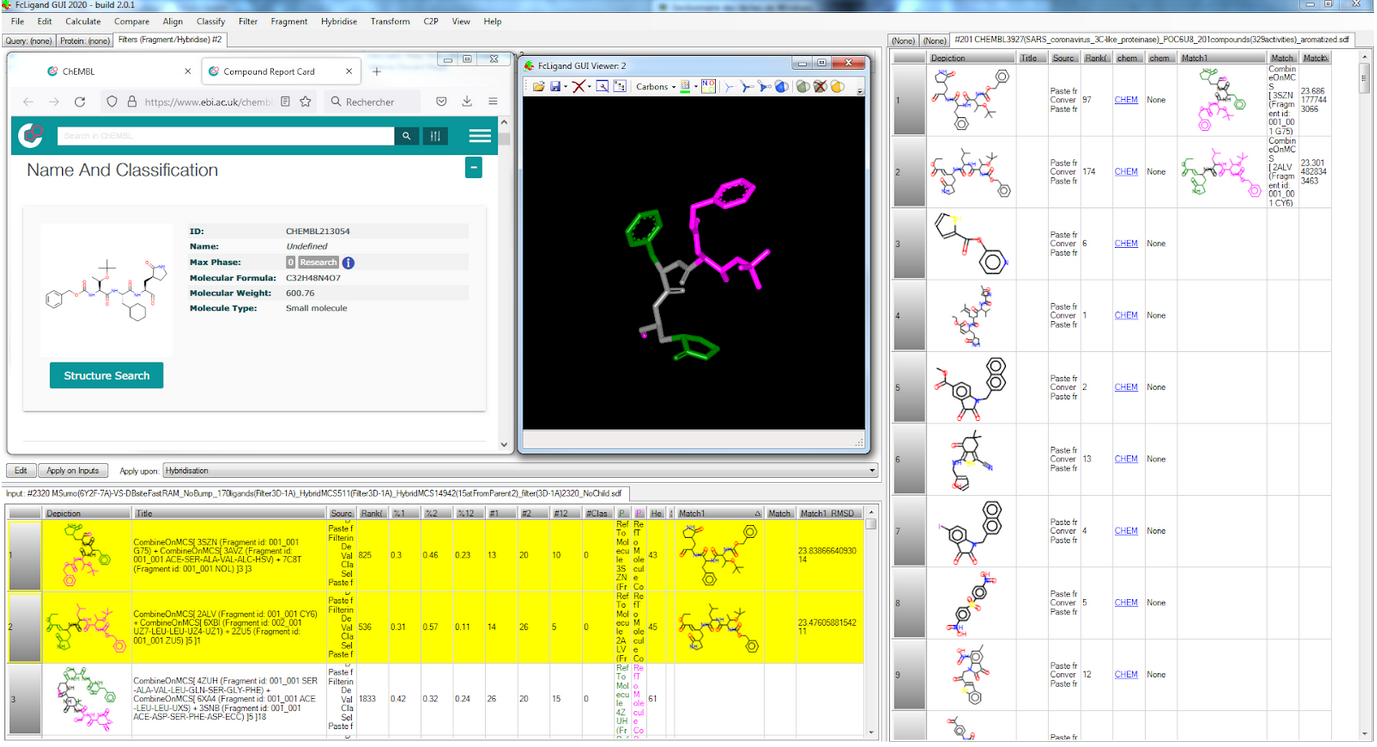
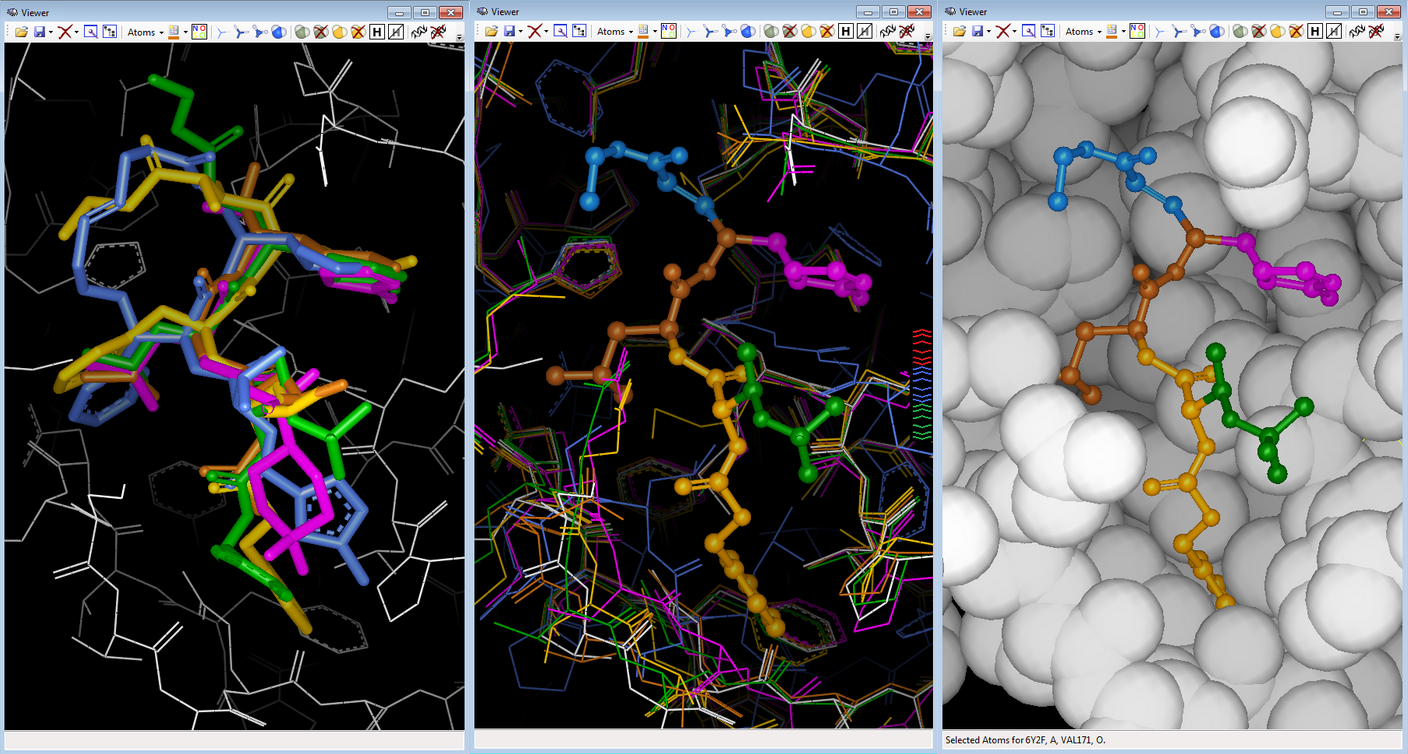
Fig. Epitope detection on SARS-CoV-2 Spike protein: spreadsheet on top summarizes 19 non-SARS-CoV-2 proteins that interact with Spike protein ; 3D viewer displays in white the 7JV4 query backbone and 3 hits in ball rendering, n°494 (7LM9 in blue), n°604 (2DD8 in orange), and n°848 (7JN5 in green vert) ; only antibody part of hits are displayed, network of 3D shared structural chemical features (Hbond donor/acceptor, charge, hydrophobicity, aromatic, …) for the 3 hits highlights putative epitope surfaces. Onto NSP 5 Main Protease (pdb code 6Y2F), we collected 209 PDB ligands having 3D similar interactions with the query and no strong steric clashes, then 3D-combined by detecting the Maximal Common Substructure to obtain 2320 hybrids ; 24 were 2D-matching to PDB ligands, 92 to Pubchem molecules, 2 to ChEMBL molecules.
 Fig. FcLigand 3D-hybrisdisation of ligand structures from 209 PDB complexes where 3D similar interaction surfaces were detected and superposed onto the 6Y2F binding site ; the displayed hybrid corresponds to CHEMBL 213054 by 3D hybridisation of 3SZN, 3AVZ and 7C8T co-crystallised ligands (shared detected scaffold is in gray)
Fig. FcLigand 3D-hybrisdisation of ligand structures from 209 PDB complexes where 3D similar interaction surfaces were detected and superposed onto the 6Y2F binding site ; the displayed hybrid corresponds to CHEMBL 213054 by 3D hybridisation of 3SZN, 3AVZ and 7C8T co-crystallised ligands (shared detected scaffold is in gray)Additionally, we used FcRebuilder on MPro to dock 212 ChEMBL active molecules on SARS-CoV-2 targets ; our software first retrieved 900 protein-ligand structures with a 3D interaction surface similar to our selected 6Y2F target. These superposed ligands were then used to 3D-rebuild the ChEMBL subset into the binding site; the predicted poses positioned at least 90% of the structure.
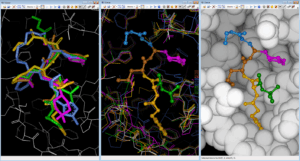 Fig. Rebuilding/Docking ChEMBL212080 into 6Y2F MPro with FcRebuilder software ; left view shows after 3D superposition into binding site five cocrystallised ligands from 7JW8 in green; 7M00 in magenta, 2A5K in yellow, 7LBN in brown, and 6LKA in blue ; middle view shows the fragment contribution to rebuild ChEMBL 212080 molecule ; right view displays the pause in the MPro target.
Fig. Rebuilding/Docking ChEMBL212080 into 6Y2F MPro with FcRebuilder software ; left view shows after 3D superposition into binding site five cocrystallised ligands from 7JW8 in green; 7M00 in magenta, 2A5K in yellow, 7LBN in brown, and 6LKA in blue ; middle view shows the fragment contribution to rebuild ChEMBL 212080 molecule ; right view displays the pause in the MPro target.These 3 POCs highlight the benefit of our PDB based drug design protocols with our software technology
-
FcLigand FcRebuilder FcBioisostere & FcTools new releases
Code standardization in C# for agile developments:
To streamline our software development processes, we decided this year to migrate some of our components to C#, our primary programming language. By bypassing dynamic serialization, we gain advantages in speed, memory usage, and enable full polymorphism for data types throughout the code.
Twenty years after it first release, C# is a now a powerful high-level programming language supported on different plateforms. At Felix Concordia, we appreciate the integration with Visual Studio IDE for faster code development, the strong typing to prevent bugs, and parallelization methods to provide high performance on multi-core processors.
We are pleased to announce the release of FcLigand 2.01, FcBioisostere 2.01, FcRebuilder 2.01, and FcTools 2.01, all in full 64-bit native architecture.
-
FcLigand v1.0.16
Major new release of the FcLigand software:
Today September 9th 2019, Felix Concordia announces the new release v1.0.16 of the FcLigand drug design software. A major effort was invested for the last two years (1) to provide a better usability with harmonized methods and tracing report per molecule, (2) to optimize most protocols with parallel mechanism, (3) to increase the overall robustness by better catching all exceptions, (4) in refactoring most fragmentation and hybridisation methods including the deliver of new amide-bond, combine-on-ring and combine-on-MCS methods, and (5) to fully update documentation.
Click here to jump to FcLigand web page
-
FcRebuilder to predict ligand bound conformation
Mining ligand common substructures in biostructures:
Today May 15th 2018, Felix Concordia announces the first release of the FcRebuilder, an alternative software solution to existing docking technologies for Drug Discovery in pharmaceuical R&D.
FcRebuilder predicts partial to full ligand 3D bound conformation in binding site(s) by mining the PDB biostructural knowledge. The Protein Data Bank having now more than 140,000 entries is an attractive source of innovation when mined at subpockets level. From pool of aligned PDB subpocket-ligand structures, FcRebuilder is extracting substructures corresponding to the ligand to predict and then 3D-combining iteratively these fragments to rebuild as much of the 2D input ligand in a bound conformation. FcRebuilder is bringing a new approach to dock molecules by taking advantage of existing biostructural knowledge.
-
a C2P-API application: Rebuilding ligand in binding site
Poster presentation at the 2017 Groupe de Graphisme & Modélisation Moléculaire conference (Reims, France):
Fast software development in drug design with the C2P-API toolkit : Rebuilding ligand bound conformation from PDB subpocket superpositions
Abstract: Literature in the FBDD field reported cases where the binding location and orientation of small co-crystallized fragments is preserved when these fragments are chemically evolved into larger ligands. Crystallography also revealed the central role of some weak fragment binders. With the benefit of the PDB growth, pocket superposition illustrated some ligand substructures having conserved binding modes towards multiple targets. Recently, we showed that 48% of 2,241 ligands structures uniquely represented in the PDB could be reproduced at 80 % of their atom coordinates within a 1.0 Å deviation by using chemical material from 3D pocket superpositions.
The development of an heuristic to rebuild ligand bound conformation from PDB pocket superpositions requires at least methods to detect Maximum Common Substructures between the compound to 3D-rebuild and all PDB ligands from superposed pockets, and methods to 3D-hybridise those MCS fragments. It was possible to develop a rebuilder prototype including a graphical interface in about 3 months by combining (a) the richness of the C2P-API Chemo-Proteomic Advanced Programming Interface, (b) the agility of the .Net C# language including reconfigurable dictionaries to include molecular rules, quasi-automatic parallelization and safe memory management, and (c) the comfort of the Microsoft Visual Studio Community IDE in developing graphical interface and debugging software.
Our auto-converging knowledge-based iterative hybridiser software is 3D-rebuilding as much as possible any input 2D-ligands into any biostructural binding site. We selected inhibitors co-crystallized in multiple PDB proteins to validate our protocol.
Click here to download the PDF poster
-
EuroQSAR-2016: poster on 3D ligand reconstruction by mining PDB
Mining PDB Subpockets to Rebuild Ligand Binding Conformations
We constituted a database of subpocket-fragment complexes (Fragmentor) through deconvolution of each PDB (Protein Data Bank) ligand in all possible fragments that match one of the chemical molecule contained in the Pubchem database. After application of a Matriochka filter, we obtained a set of 28.482 2D-fragments and 398.236 3D-fragments (PDB conformations). Subpockets were defined as protein surfaces located at 4,5 Å around fragments.
The goal of this work was to determine if there is enough information in the PDB to successfully rebuilt the binding mode of ligands, starting from the target protein structure. To test this hypothesis, we selected ligands in unique PDB entry to build a test set of 2292 protein-ligand complexes and tried to recover the position and conformation of the ligands using our Fragmentor database. We were able to predict at least 80% of the 3D-structure from 1091 ligands (48%). In conclusion, this study highlights the quality of the information contained in the PDB and supports the use of its structural information for docking tools or fragment-based drug design.
Click here to download the PDF poster
-
FcLigand v1.0.5
First release of the FcLigand software:
Today June 15th 2015, Felix Concordia announces the first release of the FcLigand, a smart software solution for Lead Generation and Lead Optimization in pharmaceuical R&D process. Designed for MS-Windows interfaces, FcLigand provides a rich set of cheminformatics methods into tabular spreadsheets. It includes among others : (1) 1D/2D/3D Filters to refine/select the set of chemical compound based on advanced filtering rules including list of unwanted chemistry, (2) 2D sub/super/MCS-structures searches, (3) Fragmentation/Deconvolution based on graph or chemistry rules to list fragments and scaffolds, (4) 3D-Hybridisation to combine in 3D pre-aligned molecules by detecting overlapping bonds, fusing rings or bonding non-overlapping pair of molecules, (5) clustering and classification on various fingerprints
FcLigand was developed upon the C2P API Advanced Programming Interface (see announcements on July 1st 2013)
Click here to jump to FcLigand web page
-
RICT-2014 conference: Poster on FcBioisostere
The 100k PDB biostructures, a unique source of Bioisosteric Replacement by Pocket Mining: further statistical analysis and validation of the FcBioisostere software solution
Bioisosteres in medicinal chemistry refers to structural changes on molecule that are not affecting existing biological activities. It’s a powerful tool to optimize the pharmaco-dynamic profile. Source of bioisosteric replacement are provided by (1) manual inspection of structure-activity literature (2) collection over such literature, (3) automatic analysis of Structure-activity data to detect chemical substitution that are maintaining the activity profile, (4) data-mining of 3D molecular data such as the CSD or the PDB databases.
Here we are exploring bioisosteric rules extracted by crossmining in 3D/2D the PDB and small Pubchem molecules to detect local pair of similar fragment-protein 3D interactions, as implemented in FC-Bioisostere software [Moriaud2011]. First, new statistics are measured on the overall distribution of those pairs of superposed PDB-based chemical moieties. 2D duplicates pairs of bioisoscteric replacement are detected and sorted according to cases where at least one pair is having the same functional annotation in the protein binding cavity, in order to score the chemical mutation suggested by other pairs having the same 2D fragments. Second structure-activitiy data related to bioisosterism mutation are qualitatively compared with FC-bioisostere pairs
This work on FC-Bioisostere explores one step further the overall chemo-proteomic challenge as initiated in our C2P Chemo-Proteomic Platform to better understand and predict interactions between, on one hand, ligands and all related fragments and, on the other hand, binding sites and all related subpockets.
-
Felix Concordia API
Pre-release of the Felix Concordia cheminformatics Advanced Programming Interface:
Today July 1st 2013, Felix Concordia is providing access to the SupBiotech engineer school (http://www.supbiotech.fr/) on an Advanced Programming Interface dedicated to cheminformatics. This set of documented informatics classes represents a consistent architecture to develop on demand new predictive methods/procedures on ligands and/or biological targets. Written in C#, this API takes advantage of the richness Microsoft .Net platform and of the agile Microsoft Visual Studio Community IDE in developing graphical interface and debugging software (Express and Community versions are accessible for free for students). Our Felix Concordia API is provided with a Doxygen-based documentation to search by keywords the required function/method/class/attribute. In such environment, Supbiotech’s students will be able to write in few days training projects in cheminformatics.
-
C2P Chemo-Proteomic initiative
The C2P Chemo-Proteomic Platform initiative to address new challenges in drug design:
On July 1st 2013, MEDIT and Felix Concordia have launched the C2P Chemo-Protein Platform initiative to provide a complete software solution to better mine altogether biostructures, structure activities and chemical libraries at PDB, Pubchem and multi-million compound levels. We strongly believe this is opening new perspectives in drug discovery and target profiling by combining knowledges from experimental data with existing predictive models.
C2P architecture allows to cross-mine with interactive 1D/2D/3D/ND queries a data repositories of biostructures, structure activities and chemical libraries. Navigation conjointly in binding sites, ligands, subpockets, fragments 2D/3D similarities including anti-targets, unwanted chemistry, and in-house data provides a new smart experience to highlight selectivity mechanisms. C2P initiative now includes MED-SuMo-GUI (Medit SA) to detect/superpose similar 3D protein surface interactions, MEDP-Fragmentor (from MEDIT) to deconvolute protein-ligand structure in pocket-fragment interactions, MEDP-Site-Classifier (Medit SA) to classify the full Protein Data Bank according to pocket similarities, FcLigand (Felix Concordia SARL) to explore 1D/2D/3D ligand similarities, FcBioisostere (Felix Concordia SARL) to better profile your compound by using bioisosteric replacement, and the C2P-API open source advanced programming interface.
MEDIT SA, a privately held software company, has developed a large experience on solutions to superpose protein binding sites for functional annotation, off-target identification, drug repurposing, and fragment-based drug design. Felix Concordia SARL is providing consistent software components to mine large set of data in life science.
-
FcBioisostere at Alabama University
First sale of FcBioisostere software:
On November 11th 2011, the Center for Biophysical Sciences and Engineering of the University of Alabama acquired multiple software licenses to use FcBioiostere on the whole campus.
FC-Bioisostere opens access to 3D bioisosteric replacement onto a lead molecule to find chemical groups having similar 3D biological interactions. While maintaining target potencies, it helps Chemists to optimize additional properties in pharmacokinetics and metabolic response, and/or to escape from existing patents by selecting alternative equivalent chemical groups.
-
FcBioisostere v1.0.4
First released of the FcBioisostere software:
Today November 1st 2011, Felix Concordia announces the first release of the FcBioisostere software, an unique technology to get access to observed equivalent chemical functions between very similar binding subpockets.
FcBioisostere is detecting pairs of very overlapping chemical substructures and storing this information in a database that can searched from the user molecule of interest. In input of FcBioisostere, we recommend to use MED-SuMo software from MEDIT SA to generate pocket and subpocket 3D-alignments from the detected superposition of a various set of features including H-bond donor, acceptor, charge, hydrophobicity, aromaticity on the binding site surface. A powerful and intuitive graphical interface in FcBioisostere allows to optimize any input 2D/3D compound by browsing and/or filtering the suggested bioisosteric pairs, and to fuse the suggested chemical substitution if your input molecule is in 3D.
FcBioisostere is bringing an new source of bioisoteric data by mining the whole Protein Data Bank, these observed equivalences should provide a net advantage for chemists having to optimize additional properties in pharmacokinetic and metabolic responses, and/or having to escape from existing patents by selecting alternative equivalent chemical groups.
-
Publication on a method to extract bioisosteres from biostructural data
A Computational Fragment Approach by Mining the Protein Data Bank: Library Design and Bioisosterism
F. Moriaud, S. A. Adcock, A. Vorotyntsev, O. Doppelt-Azeroual, S. B. Richard, and F. Delfaud*
Felix Concordia SARL, 400 av Roumanille, BP 309 06906 Sophia-Antipolis, France
MEDIT SA, 2 rue du Belvedere, 91120 Palaiseau, France
*E-mail: fdelfaud@felixc.euACS Symposium Series, In Library Design, Search Methods, and Applications of Fragment-Based Drug Design; Bienstock, R. series1076, Chapter book 5:71-88, 2011
Through database mining of the Protein Data Bank (PDB), protein pocket similarities and 3D structural alignments of similar pockets can be performed. These 3D structural alignments can serve as guides in drug design. The commercial MED-SuMo software performs superimposition of PDB ligands based on the ligand-binding corresponding pockets’ and subpockets’ 3D similarities. Subpockets are occupied by fragment-like molecules or portion of ligands. The mining of such fragments’ interaction with the macromolecule surface serves as both a target-based and fragment-based computational method for PDB mining. In this work, we describe two practical applications: (1) a ligand-based drug design technique for bioisosteric replacement and compound library design and (2) a computational fragment-based drug design protocol for target-based drug design scenarios : ligand design, ligand decoration and compound library design. The bioisosteric approach is based on a database of bioisosteric replacement rules which were dervived from the entire PDB and are applicable to any ligand with a known or predicted 3D bound conformation. We present two successful applications: the design of alkenyldiarylmethane ligands for HIV-RT, and the design of a small compound library for HSP90. A case study using the computational Fragment-Based Drug Design approach, was applied to the design of compounds for three types of protein target: Protein kinase, GPCR and kinesin.